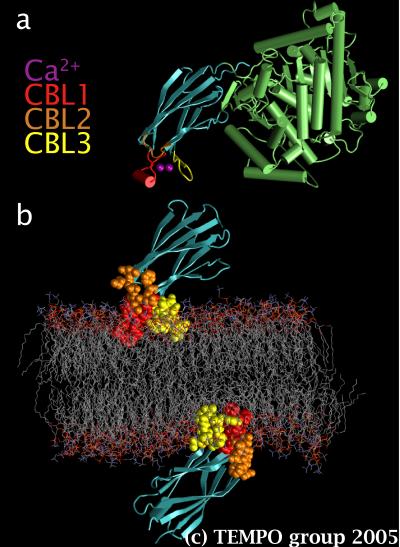
Figure 1. a) cPLA2, its 2 calcium ions, and its 3 calcium binding loops (CBL), and b) a POPC bilayer with 2 cPLA2-C2 domains.
Cytosolic phospholipase A2 (cPLA2) is an enzyme that hydrolytically liberates arachidonic acid from phosphatidylcholine (PC) in order to initiate the synthesis of leukotrienes and prostaglandin. The production of these important mediators of inflammation begins with the binding of calcium ions to the structurally distinct C2 domain of cPLA2 (cPLA2-C2), which in turn docks to PC-rich intracellular membranes. In order to inhibit this undesirable membrane targeting, the nature of the interactions between cPLA2-C2 and the phospholipids needs to be understood. Unfortunately, this information cannot be obtained experimentally because lipid bilayers are inherently so thermally agitated that it is impossible to obtain a high-resolution structure of membrane-bound proteins.
We used MD simulations to overcome this problem. cPLA2-C2 was well-suited for this approach because the crystal structure of the activated complex with two bound calcium ions is known, and because electron paramagnetic resonance (EPR) experiments show that the structure of this membrane-bound complex is similar to the crystal structure [Malmberg et al., Biochemistry 42, 13227 (2003)]. In addition, EPR measurements have defined the insertion depth and orientation in a bilayer made primarily of palmitoyl-oleyl-phosphatidylcholine (POPC). Hence, we used these results to embed two cPLA2-C2 domains on both sides of a POPC bilayer (Fig. 1.b) and run a 9 ns simulation at constant pressure and temperature (NPT) of 1 bar and 300K.
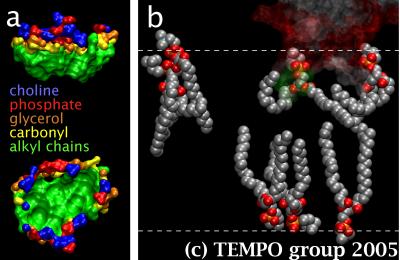
Figure2. a) Top and side views of one of he CBLs docking sites, color-coded for the different lipid subgroups constituting it, and b) a transparent C2 domain and the different types of lipids surrounding it in the system.
We found that cPLA2-C2 remodels the neighboring phospholipids to induce a docking site shaped like a hydrophobic cup with a hydrophilic rim (Fig. 2.a). C2 domains are widespread among eukaryotes. Thus, this first image ever obtained of a docking site may well be a general motif for deeply embedded peripheral membrane proteins. We discovered that the lipids next to cPLA2-C2 lost ordering after curling and wrapping themselves against the loops. This created a void under the domain, in which lipids from the opposite leaflet stretched in (Fig. 2.b). We invalidated the recent suggestion that the calcium-bridge model was applicable to this system after showing that the domain-bound calcium ions cannot contact phosphate groups from the lipids. Hence, it does not appear to be sufficient to fill the cavity between the loops to prevent cPLA2-C2 from docking.
Integral membrane proteins represent 20-30% of any given genome, and are involved in many physiological processes such as transport, signal transduction, excitation, and conduction. Because of the thermally agitated bilayer, less than one hundred high-resolution structures are known so far. Most of these, however, present a bundle of transmembrane α-helices as a basic structural motif. It is easier to obtain the sequence of amino acids of a given protein than to elucidate its structure. Therefore, it was postulated that it should be possible to predict a protein structure from its sequence by knowing the propensity of every residue in the sequence to insert into a bilayer. For a given amino acid segment, one could then sum up the contribution of each residue and determine if transmembrane insertion is favorable or not.
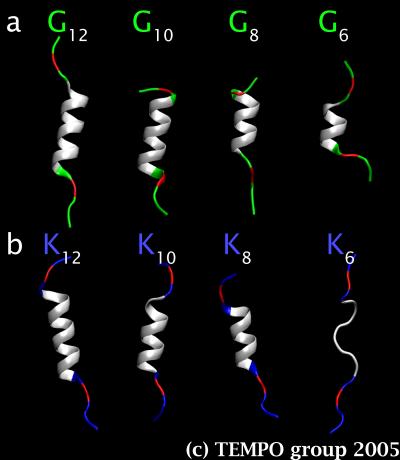
Figure 3. a) Average structures of the Gn sets, and b) of the Kn sets for n = 6 to 12.
The Wimley-White hydrophobicity scale was developed for this endeavor [Wimley et al., Nature Struct. Biol. 3, 842 (1996)]. Since the apolar core of a membrane is approximately 30 Å, one can then predict that only α-helices segments that are globally hydrophobic and that span at least 19 residues can insert, because anything shorter would force charged and polar residues (presumably located on both ends of the segment) to interact unfavorably with hydrocarbon chains of the bilayer. However, even with the small number of high-resolution structures available, it is already clear that many transmembrane segments are shorter than 19 residues, and that some are even globally hydrophilic. The failure of the simple hydrophobicity principle suggests that the solvent and/or the membrane must be playing a role in stabilizing the buried residues.
In order to test this hypothesis, we have generated two sets of hydrophobic α-helical peptides: KKPKLnKPKK (labeled Kn) and GGPGLnGPGG (labeled Gn), with n = 6, 8, 10, 12, 14, 16, 18, 20. The α-helices were made of leucine (Ln) and were capped with residues that were neither very hydrophilic nor hydrophobic, i.e. glycine and proline (GGPG), in one set, and positively charged residues, i.e. lysine (KKPK), in the other set. Every peptide was then inserted into its own POPC bilayer, and MD simulations were run to provide 10 ns of equilibrated data for every system.
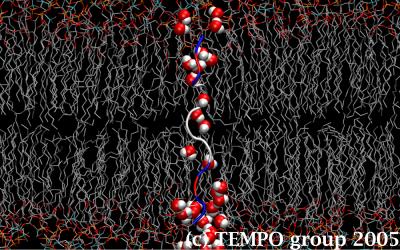
Figure 4. The K6 segment stabilizes itself by pulling in a trail of water molecules that runs through the lipid membrane.
Our results revealed that, as the helices get shorter, the capping groups make even less favorable interactions with the bilayer and hence pull down water molecules with them in order to gain energetic stability through hydrogen bonding. All of the peptides managed to retain their α-helical character, except KKPKL6KPKK (i.e. K6) (Figs. 3.a-b), which sacrificed intramolecular hydrogen bonding to intermolecular hydrogen bonding with water molecules in order for the positive capping group to maximize their interactions with bulk water and the charged lipid headgroups. The result is a trail of water molecules that made its way through the apolar core of the bilayer by sticking to the backbone of the peptide (Fig. 4). White and collaborators have performed a series of protein re-engineering and SDS-page gels experiments on the same peptides to establish their probability of insertion in the membrane as a function of the length of their α-helices. Our results have already helped explain their observations.